Past News & Events (Timeline)
December 9, 2022
In our combined experimental and computational study done in collaboration with an IOCB colleague Tomáš Slanina and his group we characterize the key intermediates of the Birch reduction process in liquid ammonia, i.e., the solvated electron, dielectron, and the benzene radical anion using two complementary approaches – synchrotron X-ray photoelectron spectroscopy in liquid microjets and cyclic voltammetry. First, this has allowed us to extract mutually consistent values of electron binding energies/redox potentials of these species, whichare in quantitative agreement with our electronic structure calculations. Second, this case study showed what are the pitfalls when attempting to bridge the two experimental techniques and how careful one has to be when relating results of photoelectron spectroscopy measured with respect to the vacuum level to cyclic voltammetry employing a standard electrode.
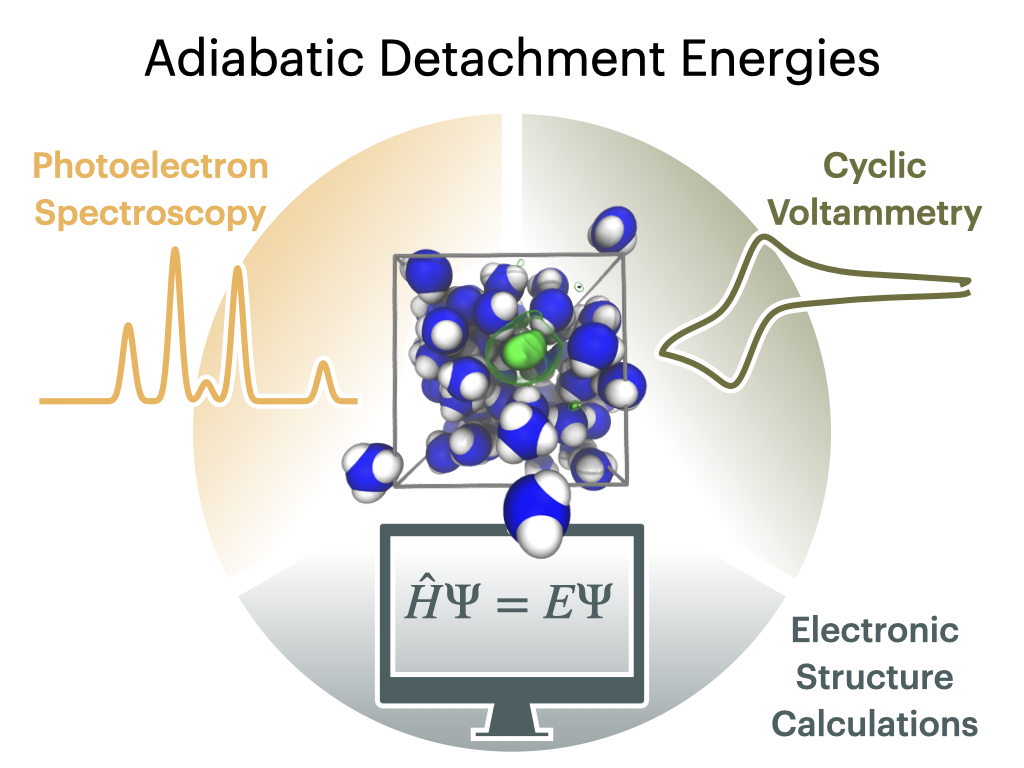
Nemirovich T., Košťál V., Copko J., Schewe H.Ch., Boháčová S., Martinek T., Slanina T., Jungwirth P.:
Bridging Electrochemistry and Photoelectron Spectroscopy in the Context of Birch Reduction: Detachment Energies and Redox Potentials of Electron, Dielectron, and Benzene Radical Anion in Liquid Ammonia.
Journal of the American Chemical Society 144 (48): 22093–22100, 2022.
August 3, 2022
In our JPCB Perspective Article we propose with our collaborators in Finland, Norway, and Germany a practical path employing automated parameterization tools to systematically building up physically well-justifed models for biomembrane systems in native aqueous environments, which effectively account for electronic polarization via charge scaling. This work has been already initiated within the open collaboration NMRlipids project (nmrlipids.blogspot.com) and benefits from creation of databanks of molecular dynamics simulations with enforced quality control. The ultimate goal is to provide the scientific community a consistent charge-scaled biomolecular force field allowing to obtain in a computationally efficient way "the right results for the right reasons" and you are most welcome to participate on this collaborative project.
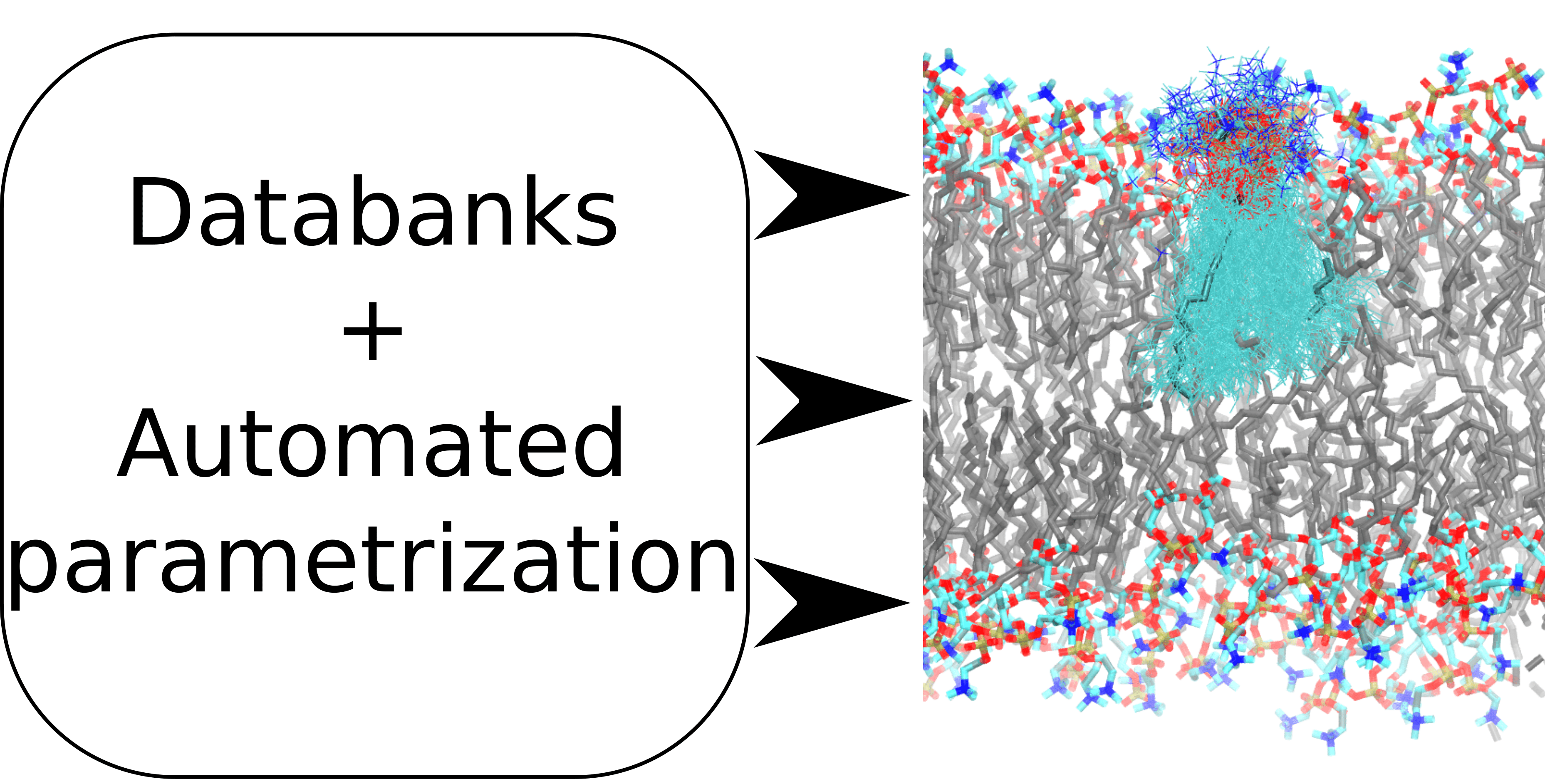
Antila H.S., Kav B., Miettinen M.S., Martinez-Seara H., Jungwirth P., Samuli Ollila O.H.:
Emerging Era of Biomolecular Membrane Simulations: Automated Physically-Justified Force Field Development and Quality-Evaluated Databanks.
Journal of Physical Chemistry B 126: 4169, 2022.
July 29, 2021
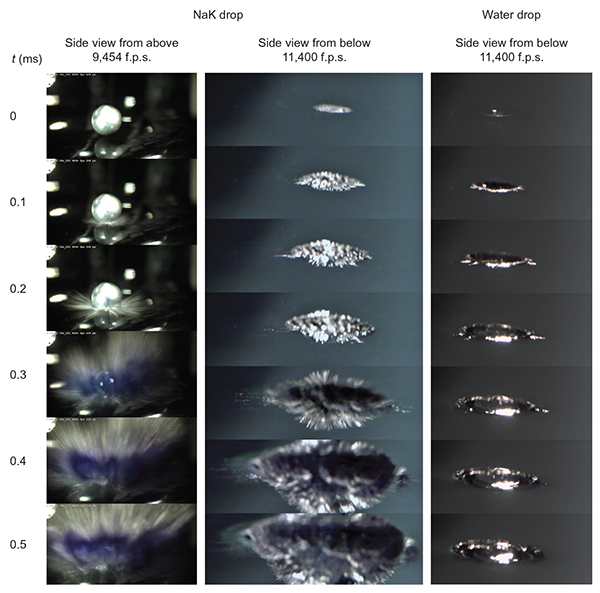
The idea of using high pressure to make metal out of water is nothing new. However, the required pressure of 50 Mbar may exist in the cores of large planets, but not at terrestrial conditions. In collaboration with colleagues and friends (thank you Steve, Bernd, Robert, Tillmann,…) from the University of Southern California, the Fritz Haber Institute, the BESSY II synchrotron, and other institutes, our group (thank you Phil, Christian, Vojta, Marco,…) developed a method that allowed us to a prepare metallic water solution while completely sidestepping the need for high pressure. Inspired by work with alkali metal-liquid ammonia solutions, which at high concentrations behave like a metal, we decided to attempt formation of a conduction band not by compressing water molecules but rather by massive dissolution of electrons released from the alkali metal. In doing so, however, we had to overcome a fundamental obstacle, namely that on introduction to water, alkali metals tend to explode.
The successful way around the explosive chemistry was in exposing (inside a vacuum chamber) a drop of liquid sodium-potassium alloy to a small amount of water vapor, which began to condense on its surface. Electrons liberated from the alkali metal dissolved in the layer of water on the surface of the drop faster than the chemical reaction that results in the explosion proceeds. Moreover, there was a sufficient number of them to overcome the critical limit for the formation of a conduction band, giving thus rise to a beautiful golden metallic water solution, which we then characterized using optical reflectance and synchrotron X-ray photoelectron spectroscopies.

Mason P.E., Schewe Ch.H., Buttersack T., Košťál V., Vitek M., McMullen R.S., Ali H., Trinter F., Lee Ch., Neumark D.M., Thürmer S., Seidel R., Winter B., Bradforth S.E., Jungwirth P.:
Spectroscopic Evidence for a Gold-Coloured Metallic Water Solution.
Nature 595: 673, 2021.
April 6, 2021
Ordinary pure water has no distinct taste, but how about heavy water – does it taste sweet, as anecdotal evidence going back to 1930s may have indicated? And if yes – why, when D2O is chemically practically identical to H2O, of which it is a stable naturally-occurring isotope? These questions arose shortly after heavy water was isolated almost 100 years ago, but they had not been satisfactorily answered until now. Together with the group of Masha Niv at the Hebrew University and Maik Behrens at the Technical University of Munich, we have now found answers to these questions using molecular dynamics simulations, cell-based experiments, mouse models, and human subjects. In our article published in Communications Biology, we show conclusively that, unlike ordinary water, heavy water tastes mildly sweet to humans but not to mice, with this effect being mediated by the human sweet taste receptor. But why does it taste sweet and not, e.g., salty or bitter, remains to be established...

Abu N.B., Mason P.E., Klein H., Dubovski N., Shoshan-Galeczki Y.B., Malach E., Pražienková V., Maletínská L., Tempra C., Chamorro V.C., Cvačka J., Behrens M., Niv M.Y., Jungwirth P.:
Sweet taste of heavy water.
Communications Biology 4: 440, 2021.
April 1, 2021
This may look like April Fools but it's actually a serious study where we used molecular simulations with charge scaling to resolve the following puzzle - why are the experimental number densities of aqueous solutions of LiCl and NaCl practically the same at equal concentrations? Normally, one would assume that the number density of NaCl(aq) should be lower than that of LiCl(aq) since sodium cationsare larger than lithium cations. The trick is that lithium tightly orients four H2O molecules in its first solvation shell, such that these waters cannot make good hydrogen bonds with neighboring molecules. As a result, voids occur in the solvation environment of Li+ which compensate the tight first solvation shell, yielding an overall number density equal to the more regular sodium chloride solution.

Nguyen M.T.H., Ticháček O., Martinez-Seara H., Mason P.E., Jungwirth P.:
Resolving the Equal Number Density Puzzle: Molecular Picture from Simulations of LiCl(aq) and NaCl(aq).
Journal of Physical Chemistry B 125: 3153, 2021.
August 13, 2020
JCP just published our review article where we provide a practical guide how to effectively account for electronic polarization in molecular simulations via charge scaling. Bottom line - the future of charge scaling is bright but there is a lot of work still in front of us!

Duboué-Dijon E., Javanainen M., Delcroix P., Jungwirth P., Martinez-Seara H.:
A practical guide to biologically-relevant molecular simulations with charge scaling for electronic polarization.
Journal of Chemical Physics 153: 050901, 2020.
June 5, 2020
How is a metal formed? With colleagues from the Fritz-Haber Institute, the Berlin synchrotron BESSY II, USC Los Angeles, and Charles University in Prague we used liquid microjet photoelectron spectroscopy and ab initio molecular dynamics to map at the molecular level the electrolyte-to-metal transition in alkali metal–liquid ammonia solutions. In other words we followed in detail how the excess electrons liberated from the alkali metal change from blue localized solvated electrons to delocalized species rendering the solution a bronze-gold metallic sheen. Watching these spectacular solutions one gratefully realizes that science cane be not only exciting but also beautiful.

Buttersack T., Mason P.E., McMullen R.S., Schewe C., Martínek T., Březina K., Crhan M., Gomez A., Hein D., Wartner G., Seidel R., Ali H., Thurmer S., Maršálek O., Winter B., Bradforth S.E., Jungwirth P.:
Photoelectron spectra of alkali metal–ammonia microjets: From blue electrolyte to bronze metal.
Science 368 (6495): 1086-1091, 2020.
April 21, 2020

April 12, 2020
Review of Scientific Instruments has published an Editorial Highlight (which they call Scilight) about our article therein. This paper summarizes our development of a refrigerated liquid ammonia microjet apparatus which we then used with our Berlin and USC colleagues to measure at the BESSY II synchrotron photoelectron spectra of alkali metal - liquid ammonia solutions (to be published - stay tuned). This is what happens if you give a theoretical group a bit of lab space...

December 12, 2019
Congratulations to the winners of the 2019 Martina Roeselova Memorial Fellowship!
With the help of all the private donors and IOCB Tech we are happy to support four bright young scientists/parents and their families - Lenka Ťupová from the Charles University, Faculty of Pharmacy in Hradec Králové, Jaroslava Nováková from the Charles University, Faculty of Mathematics and Physics in Prague, Aneta Ledererová from the Masaryk University, Faculty of Medicine in Brno, and Miroslav Dvorský (first ever father to be awarded!) from the Institute of Botany of the Academy in Třeboň. Big thanks to all who contributed with their money, time, and advice!

December 5, 2019
There are no free lunches...well, maybe except when including electronic polarization effects into nominally non-polarizable force field molecular dynamics simulations via charge scaling. Feel free to taste this free lunch and have a look at our Charge Scaling Manifesto, where we outline with Brian Kirby from Cornell University the basic physics behind charge scaling and point to recent successes and pitfalls. And of course feel free to join the ever growing crowd of charge scaling simulators!

December 3, 2019
Congratulations to the winners of the Dream Chemistry Award 2019!
This year's TOP 5 Dream Chemistry Awards go to Yujia Qing (Oxford U) - who is the absolute winner of the competition organized jointly by IOCB Prague and IPC Warsaw, as well as to Hannes Mikula (TU Wien), Jeffrey Martell (U Wisconsin), Yoeri van de Burgt (TU Eindhoven), and Emiliano Cortes (LMU Munich). Congrats to all and big thanks to Bara and her team for organization, to Robert and all jury members for evaluation work, and to IOCB and IOCB Tech managment for support!


April 9, 2019
Our group is inspecting the newly installed computer cluster - the biggest in IOCB so far with over 240 nodes - all fresh from the owen. Thank you very much European Union for grant support of 2 M € from the Structural Funds!

March 9, 2019
Thanks to everybody for making our annual group seminar/teambuilding at the top of the highest Czech mountain Sněžka a success - meaning we did not loose anybody despite the hurricane up there :). When we passed the group with Tibetian flags commemorating the 60th anniversary of the failed uprising against Chinese rule it really felt like we were in the Himalayas!

December 17, 2018
Big congratulations to the four winners of the 2018 Martina Roeselová Memorial Fellowship!
Support to young scientists raising small kids goes to Sylvie Rimpelová, Barbora Melkes, Petra Šedová, and Klára Frydrýsková! The award ceremony at the Christmas party of the Institute on December 14 was great fun and big thanks to all that helped - the small donors, the Institute and IOCB Tech, member of the selection comittee, and the Memorial Fund!

December 10, 2018
Last year around this time we presented here an artistic Christmas movie showing the beauties of solvated electrons in ammonia. Well, now we've actually managed to measure their photoelectron spectra in a cool liquid ammonia microjet going from the blue regime of (di)electrons all the way to the golden/bronze metallic state. This has been a massive collective effort together with our buddies - Steve Bradforth's group at USC, Bernd Winter's group at the Fritz Haber Institute in Berlin, and Robert Seidel and his people at the BESSY synchrotron. Big thanks to everybody on the picture, which also shows the first spotting of the photoelectron signature of an ammoniated electron! Turns out this is a shy nocturnal animal, which usually shows up only late at night and stinks a lot...

November 8, 2018
Our latest study on cell penetrating peptides, combining molecular simulations with fluorescence and electron microscopy of model vesicles and cells, has just been published in PNAS. After a long march through journals (thank you Russell in Nature Chemistry and all the critical anonymous reviewers for helping to make the study better) we are coming out with a suggestion that the passive cell penetration mechanism of arginine-rich peptides may not be a simple opening of a transition pore but rather involves complex membrane bifurcation and fusion processes, the latter being analogous to the well known calcium induced membrane fusion. Passive cell penetration and membrane fusion may thus be two sides of the same coin!
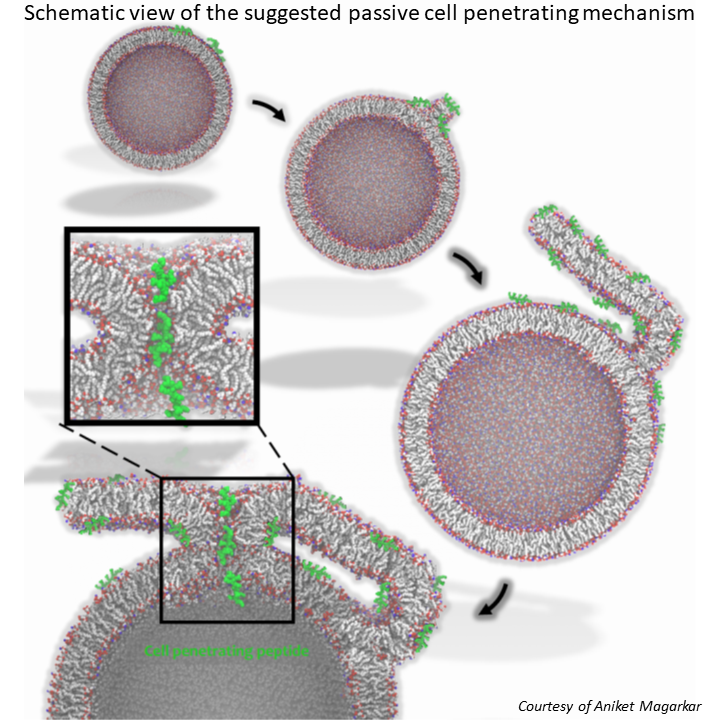
October 29, 2018
Happy birthday JACS!
JACS is turning 140 and as a part of the birthday celebration the editors selected representative papers from recent years. We are happy that our colleague Paul Cremer selected our joint paper dealing with the effect of guanidinium on stability of boiomacromolecules for the year 2017. Thanks Paul!

May 7, 2018
Big congratulations and thanks to everybody on the picture, in particular to Tillmann and Phil, for producing after a year of work in the lab the first ever liquid ammonia microjet! Not a simple task given the fact that it all has to be cooled down to about -50 °C. This allows us to measure at the Berlin synchrotron BESSY II the photoelectron spectra of liquid ammonia and hopefully eventually to characterize the solvated electrons therein, in what should become a new twist of our never-ending "throwing sodium into something" story.

March 6, 2018
The American Institute of Physics has highlighted in an editorial our new article in the Journal of Chemical Physics on developing a simple but accurate model of calcium for biological simulations. Sweet, since this is certainly not a breakthrough discovery but rather an incremental results of a tedious down-to-the-earth work, which will hopefully be useful to the community. If you are modeling calcium signaling or any other process involving this lovely but difficult ion, feel free to use our new parameterization and let us know how it works for you!
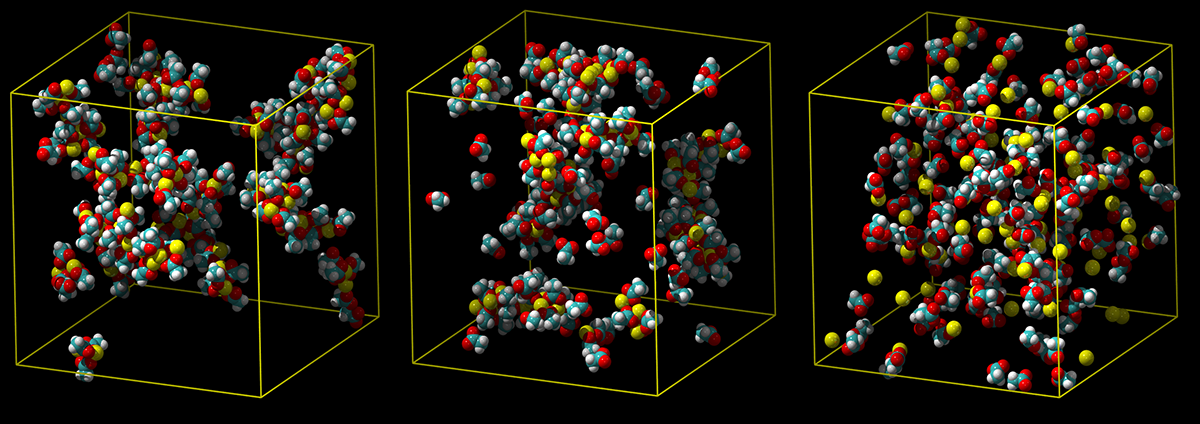
A standard calcium model overestimates how strongly calcium binds, leading to clumps of ion pairs (left). An intermediate model shows less clumping (middle),
and a refined charge-scale model correctly predicts a weak association with carboxylic groups in water (not shown) (right).
Martinek T., Duboué-Dijon E., Timr Š., Mason P.E., Baxová K., Fischer H.E., Schmidt B., Pluhařová E., Jungwirth P.:
Calcium Ions in Aqueous Solutions: Accurate Force Field Description Aided by Ab Initio Molecular Dynamics and Neutron Scattering.
Journal of Chemical Physics 148 : 222813 (2018).
February 23, 2018
Congratulations Ondra!
Ondřej Ticháček got for his work on a computational model of the inner ear and the auditory nerve the Werner von Siemens prize for the third best master thesis in sciences and engineering in 2017. It was a fun party, we both dressed sharp, got a free dinner and a tiny part of the whole sum.

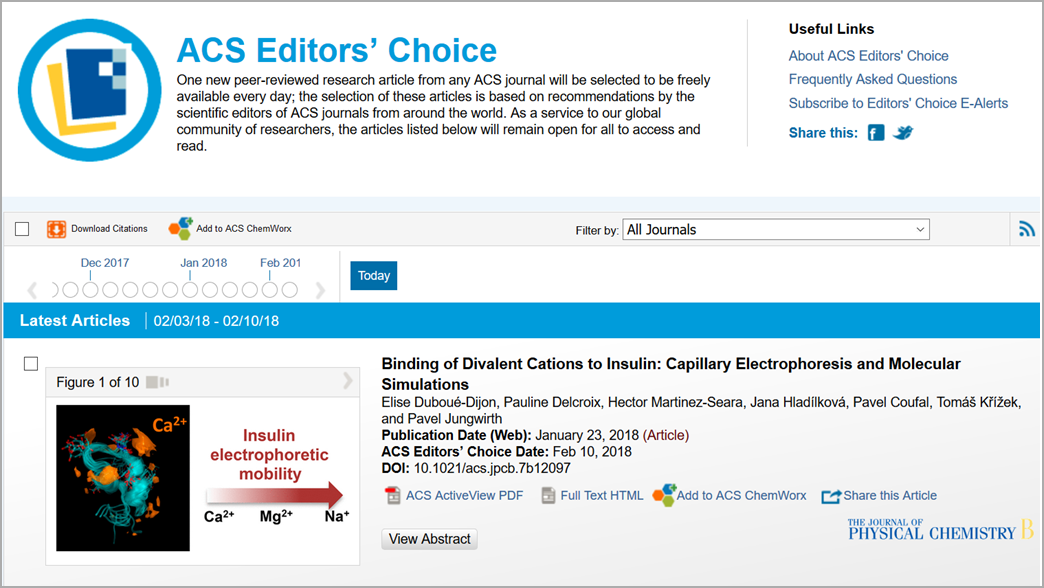
February 10, 2018
Our latest JPC B article on interactions of divalent cations with insulin has been selected as Editor's Choice among papers published in all ACS journals. Possibly, the editors were as intrigued as we were by the disagreement between our results obtained by capillary electrophoresis vs. those from our molecular dynamics simulations. Or, maybe, they liked our hand-waving argument potentially blaming restructuring and/or aggregation of insulin molecules upon ion binding for the disagreement.
January 5, 2018
Throughout the year 2017, I participated, as (maybe) an expert on memory of water, on a Statement on homeopathy by the European Academies Science Advisory Council (EASAC). The whole report with a typical Brussels language title "Homeopatic products and practices: assessing the evidence and ensuring consistency in regulating medical claims in the EU" can be downloaded here. The common language title of our report may be something like "Hey guys, do not even try to use the memory-of-water explanation, homeopathy is placebo at best, so why don't you call it like this?". We are far from calling for a ban on homeopatic products since we fully respect the freedom of customers choice. At the same time, patients have the right for evidence-based information and should be protected from false advertisement. Therefore, we make the following recommendations (citing from our report):
- There should be consistent regulatory requirements to demonstrate efficacy, safety and quality of all products for human and veterinary medicine, to be based on verifiable and objective evidence, commensurate with the nature of the claims being made. In the absence of this evidence, a product should be neither approvable nor registrable by national regulatory agencies for the designation medicinal product.
- Evidence-based public health systems should not reimburse homeopathic products and practices unless they are demonstrated to be efficacious and safe by rigorous testing.
- The composition of homeopathic remedies should be labelled in a similar way to other health products available: that is, there should be an accurate, clear and simple description of the ingredients and their amounts present in the formulation.
- Advertising and marketing of homeopathic products and services must conform to established standards of accuracy and clarity. Promotional claims for efficacy, safety and quality should not be made without demonstrable and reproducible evidence.

|
|
December 31, 2017 - All good in 2018 from solvated electrons!
December 15, 2017 - Congratulations to winners of the 2017 Martina Roeselová Memorial Fellowship

This year fellowship aimed at supporting Ph.D. students and postdocs raising small kids goes to Barbora Planková, Kateřina Sam, Lucie Pokorná, Adéla Melcrová, and their families.
Big thanks to all generous donors and members of the selection comittee!
www.nfmr.cz
December 15, 2017
 |
Happy Ph.D. graduation, Štěpán!We will miss you, but know you will do great as a postdoc in Paris... |
December 5, 2017 - Congratulations to the winner and finalists of the 2017 Dream Chemistry Award

|
This year winner of the Dream Chemistry Award is Jessica Kramer from the Univeristy of Utah. The remaining finalists are Justin Chalker (Flinders University),
Rob Ameloot (KU Leuven), Nathan Crook (Washington University), and Yogesh Surendranath (MIT). Thank you Bára, Dušan, Robert, members of the jury, IOCB, and TTO for help and support!
|

October 2017
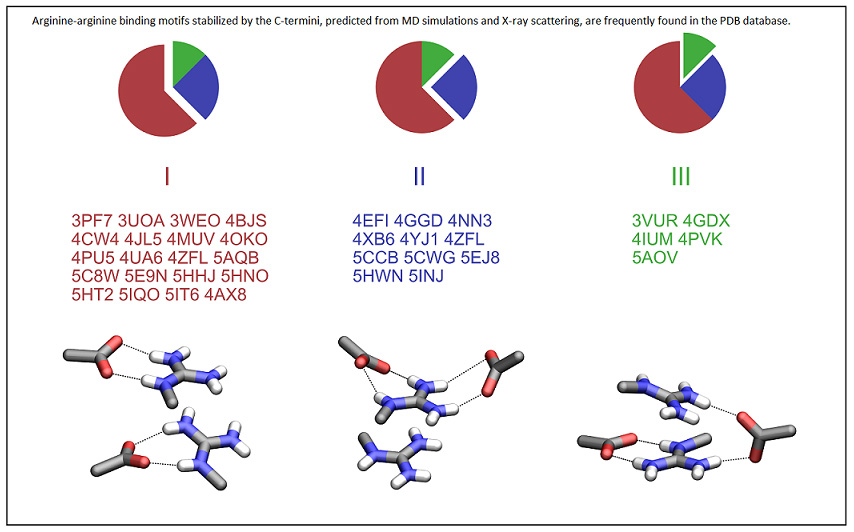
PNAS has just published our discovery of a binding motif which holds together arginine-rich cationic peptides. We know from school that ions or charged groups carrying a charge of the same sign repel each other. In aqueous media, however, this electrostatic repulsion is significantly reduced and can be compensated by other molecular forces. Using molecular dynamics and X-ray scattering our international team encompassing Lund, Prague, Grenoble, and Zagreb, unraveled a new binding pattern, wherein the positively charged side chains of arginine amino acids bind to each other in the presence of negatively charged terminal groups or side chains of peptides or proteins. Subsequently, we have identified an analogous motif in more than two hundred biologically significant structures in the protein database. Our findings provide the key to the understanding of the self-association mechanism in oligo-arginines with implications for ther potential biological roles including a cell penetration ability.
July 2017
In the role of a Journal of Physical Chemistry editor
We have just published with my colleague-editor Arun Yethiraj a viewpoint on how we think a suitable manuscript for JPC on biomolecular or polymer simulation should look like.
Maybe you'll find this useful or at least amusing...

March 2017
We have published with our experimental and computational colleagues our best attempt to summarize what we know about the Hofmeister series today. In the last decade, we have been obsessed with ion-specific (Hofmeister) effects on proteins and this Feature Article may be viewed as a sort of therapy converting an unhealthy obsession into a molecular understanding.

August 2016
Angewadte Chemie (A Non-Exploding Alkali Metal Drop on Water: From Blue Solvated Electrons to Bursting Molten Hydroxide) has selected the continuation of our "balcony experiments" with alkali metals in water for inside cover (below). As Phil says: "Explosions are sooo much last year science," so this year we followed the non-explosive (but vigorous) reaction of a sodium/potassium alloy drop gently placed on water. And with our colleagues from Braunschweig - Sigurd and Tillmann, we saw amazing chemistry happening. This includes blue solvated electrons visible with a naked eye despite their ephemeral lifetime, colorful evaporation of the alkali metals, a burning red drop, ... and, amazingly, the final "transmutation"of the metal drop into a transparent "marble" of molten hydroxide supported at the water surface via the Leidenfrost effect (same as that stabilizing water drops on a hot stove or that allowing you (not me!) to walk on hot ashes). Words cannot describe the beauty of this in full so go ahead and check our YouTube video.
The story has been also covered in the popular science literature, see: Chemistry World and New Scientist

January 2016
Chemistry World of the Royal Society of Chemistry selected our study of the mechanism of alkali metal explosions in water in their editorial Cutting edge chemistry in 2015 under a rather appropriately chosen title "Back to school" ☺
July 2015
The Journal of Physical Chemistry Letters has published a Viewpoint of mine entitled Biological water or rather water in biology? (JPCL 6: 2449–2451, 2015). The key message, summarized in a somewhat lighter tone in my amateurish drawing below, is as follows: While water with dissolved ions and osmolytes is essential for establishing homeostasis, it is primarily the biomolecule itself which carries the biological function. It is perfectly justifiable to talk about water in biology and discuss the role of interfacial water around biomolecules with its distinct properties. However, I would argue that the often used term "biological water", with all its connotations toward a hypothetical state of cellular "vicinal water" carrying biological function, is bringing us dangerously close to the long obsolete concept of "vis vitalis" and should, therefore, be dropped.
January 2015
Nature Chemistry published on January 26, 2015 results of our "balcony experiments" on alkali metal explosions in water (see: Coulomb explosion during the early stages of the reaction of alkali metals with water). This is NOT what we normally do, but we love the blasts (especially Phil does!) and we want to understand why the thing explodes. Because it should not - the steam and hydrogen gas produced at the interface between the alkali metal and water should effectively separate the two reactants quenching thus the reaction. So, why the hell DOES it explode? Using high speed cameras, molecular dynamics simulations, and back-of-the-envelope calculations we found out the the key piece missing in the puzzle is a Coulomb explosion of the metal prior to the steam and hydrogen blast. As electrons leave the metal for water (to react there creating hydrogen and hydroxide) the metal charges positively to the extent that it becomes unstable (so called Rayleigh instability, same as in electrospray) and shoots out metal spikes into water, ensuring thus efficient mixing of reactants and enabling the explosive behavior.
There was also a minor explosion in the popular science literature following our blast, see e.g.:
January 2015
The editors of the January 14, 2015 issue of JACS chose as the cover ilustration our picture showing in an artistic way ionization of aqueous DNA by synchrotron radiation and the corresponsing ionization energies of the individual aqueous DNA bases. In the words of the Editor: "Ionizing radiation can induce oxidative damage to DNA, including double-strand breaks, which can lead to mutations and possibly cancer. To theoretically predict the rate of certain indicators of this damage, researchers need several pieces of information, including the one-electron redox potential of the five nucleobases. However, it has been difficult to measure these properties electrochemically, and other recent approaches have not considered the bases in aqueous solution, their native habitat. Now Petr Slavíček, Bernd Winter, Pavel Jungwirth, Stephen E. Bradforth and colleagues have determined the one-electron redox potentials of nucleic acid bases in water with a combined experimental-computational approach.They use liquid-jet photoelectron spectroscopy to measure the vertical ionization energies of nucleobase components, including purine and pyrimidine nucleotides, nucleosides, pentose sugars, and inorganic phosphate, then use quantum chemistry calculations to estimate the reorganization energies to the change in nucleobase charge. By combining these two data sets, the authors have determined sufficiently accurate reduction potentials for nucleobases in their native environment; values that could help to clarify the rate of oxidative damage to DNA in sunlight and higher-energy sources of radiation."
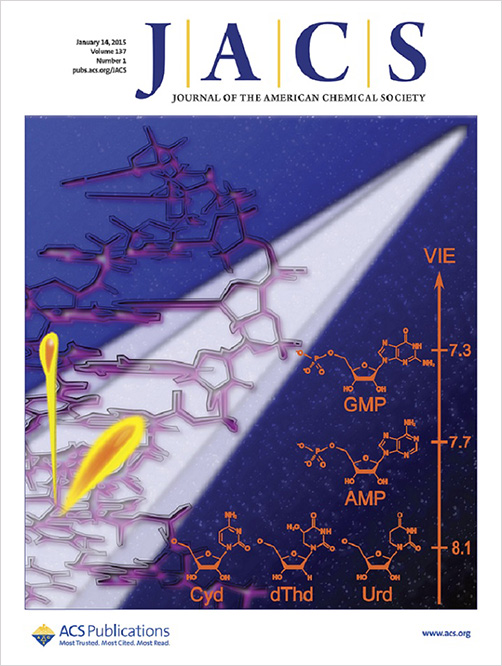
Pluhařová E., Schroeder C., Seidel R., Bradforth S.E., Winter B., Faubel M., Slavíček P., Jungwirth P.:
Oxidation Half-Reaction of Aqueous Nucleosides and Nucleotides via Photoelectron Spectroscopy Augmented by Ab Initio Calculations.
Journal of the American Chemical Society 137: 201 (2015).
March 2014
The editors of the March 2014 issue of Electrophoresis (Special Issue on Fundamentals) picked for the cover our figure showing electrophoretic mobilities of neutral markers in aqueous salt solutions. In the related article we demonstrate using capillary electrophoresis and molecular dynamics simulations two things: i) that neutral markers can have non-zero electrophoretic mobilities thanks to specific interactions with salt ions from the solution and ii) quantitatively, these mobilities depend on the chemical composition of a particular marker. As a result, we show that there is no "perfect" neutral electrophoretic marker, since any marker can acquire a non-zero mobility due to interactions with dissolved ions. This mobility is typically small, but clearly measurable.

Křížek T., Kubíčková A., Hladílková J., Coufal P., Heyda J., Jungwirth P.:
Electrophoretic mobilities of neutral analytes and markers in aqueous solutions of Hofmeister salts.
Electrophoresis 35: 617-624 (2014).
January 2014
A cover on the January 30, 2014 issue of the Journal of Physical Chemistry B shows our most recent joint experimental and computational study aimed at elucidating the orientational distribution of a fluorescence dye in a model phospholipid membrane. Combination of ab initio and molecular dynamics calculations yeilded a distribution which is in a very good agreement with the result obtained from fluorescence detected linear dichroism. The ultimate goal is to develop our approaches into a technique capable of yielding detailed quantitative structural information on membrane proteins in living cells.

Timr Š., Bondar A., Cwiklik L., Štefl M., Hof M., Vazdar M., Lazar J., Jungwirth P.:
Accurate Determination of the Orientational Distribution of a Fluorescent Molecule in a Phospholipid Membrane.
Journal of Physical Chemistry B 118: 855 (2014).
November 2012
Science has covered our recent JPCL paper on the structure of the hydrated electron as a short article in the section Editor's Choice. This paper entitled Unraveling the Complex Nature of the Hydrated Electron presents ab initio molecular dynamics simulations of an electron in liquid water performed by my students Frank Uhlig and Ondrej Marsalek. As the picture below suggests we indeed got the hydrated electron under a (quantum mechanical) magnifying glass!
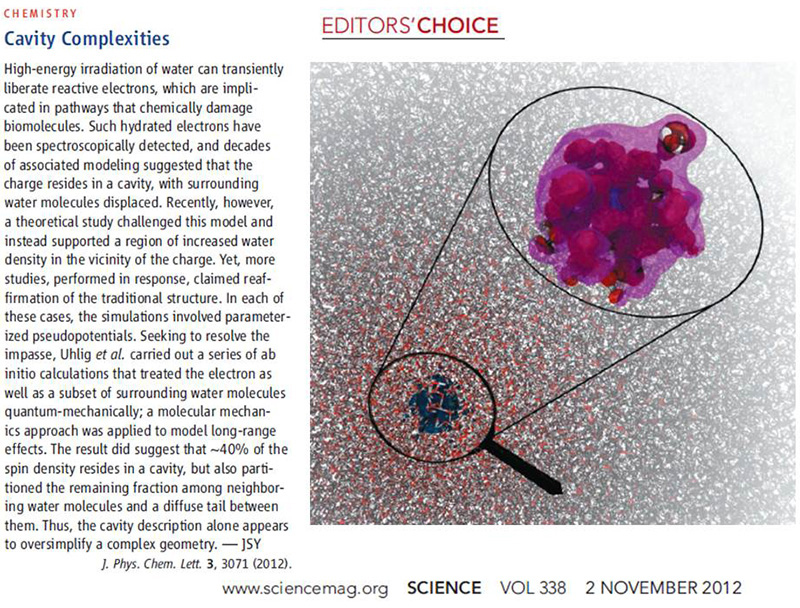
Uhlig F., Maršálek O., Jungwirth P.:
Unraveling the Complex Nature of the Hydrated Electron.
Journal of Physical Chemistry Letters 3: 3071 (2012).
July 2012
Chemical & Engineering News recently published an article about Hofmeister effects showing results from our JACS paper with Paul Cremer's and Christian Hilty's groups at Texas A&M University.

Rembert K., Paterová J., Heyda J., Hilty C., Jungwirth P., Cremer P.S.:
The Molecular Mechanisms of Ion-Specific Effects on Proteins.
Journal of the American Chemical Society 134: 10039 (2012).
June 2011
Victoria Buch Memorial Issue: Below is the cover of the special JPCA issue in memory of late Victoria Buch (1954-2009) put together by her colleagues. Check this issue out and remember Victoria as a great scientist, warm friend, and passionate human rights activist!

January 2010
Reaction of a proton and an electron toward a hydrogen atom is the simplest chemical process I can think of. It becomes, however, much more intriguing if it is happening in water. With Ondřej Maršálek in Prague, Tomaso Frigato and Burkhard Schmidt in Berlin, Joost VandeVondele in Zurich, and Steve Bradforth at USC we were able to capture using ab initio dynamics the molecular mechanism of this process with gory molecular detail. In agreement with kinetic measurements we showed that the process is a proton transfer (and not electron transfer) reaction, which is fast but not diffusion limited. The former is true since proton has a lower effective mass in water (it is "lighter") than hydrated electron. The latter is due to solvation effects. Namely, the two charged particles (i.e., proton and electron) have to first shed off their solvent shells before they can form the neutral hydrogen atom. The cost of this is almost as large as the binding energy of an H atom. The below journal cover shows the rection path by which a proton moves in a water cluster to a hydrated electron to form a hydrogen atom.

Maršálek O., Frigato T., VandeVondele J., Bradforth S.E., Schmidt B., Schuette C., Jungwirth P.:
Hydrogen forms in water by proton transfer to a distorted electron.
Journal of Physical Chemistry B 114: 915-920 (2010).
July 2009
Pairing between like-charged side chains in polyarginine. We have shown that the guanidinium cations forming arginine side chains tend to pair in water despite the obnious Couloumb repulsion between them. This work builds on previous work of other groups who showed that guanidinium forms contact ion pairs in aqueous salt solutions. The present combined MD simulations and ab initio PCM calculations also allow us to trace this effect to a favorable combination of electrostatic, dispersion, and cavitation effects for the disc-shaped, quasi-aromatic guanidinium ions with an inhomogeneous internal distribution of charge. Analysis using the Protein Data Bank shows that such an associative behavior of arginine occurs frequently within (as well as inbetween) proteins with potential implications for enzymatic activities and protein association patterns. The below journal covers graphically depict side chain pairing in polyarginine and lack thereof in a control simulation of polylysine.

Vondrášek J., Mason P.E., Heyda J., Collins K.D., Jungwirth P.:
The Molecular Origin of Like-Charge Arginine-Arginine Pairing in Water.
Journal of Physical Chemistry B 113: 9041-9045 (2009).
June & July 2008
Unraveling ionization processes in water and of aqueous biomolecules connected with indirect and direct ionization damage. With our colleagues Steve Bardforth and Anna Krylov (USC), Bernd Winter (BESSY Berlin), Tomaso Frigato and Burkhard Schmidt (FU Berlin) and Petr Slavíček (Inst. of Chem. Technol. Prague) we are investigating ionization processes in water, as well as for aqueous DNA components and side chain models of amino acids. Within the former, we follow the fate of the cationic hole in water (leading to H3O⊕ and OH) and the photodetached electron (leading to solvated electron). Within the latter, we are establishing vertical ionization potentials of aqueous DNA bases, nucleosides, nucleotides, and titratable side-chain groups of amino acids. We are combining ab initio calculations, DFT-based ab initio molecular dynamics, and methods employing a non-equilibrium polarizable continuum model to relate to photoelectron spectroscopy measurements. The below journal covers graphically depict ionization in aqueous protonated imidazole and the proton-transfer dynamics of the cationic hole in a water dimer.
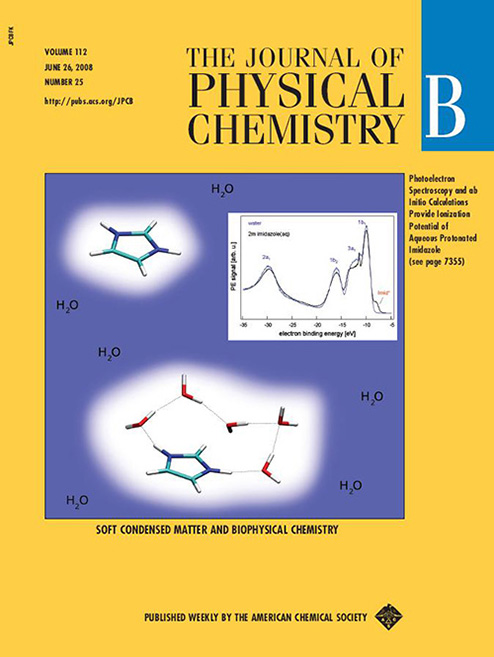
Jagoda-Cwiklik B., Slavíček P., Noltig D., Winter B., Jungwirth P.: |

Frigato T., VandeVondele J., Schmidt B., Schuette C., Jungwirth P.: |
May 2008
"Filming" ice nucleation and freezing in pure & salty water by simulation & experiment. With our German and Israeli colleagues Sigurd Bauerecker and Victoria Buch we have developed a concept of computational and experimental filming of freezing. On the experimental side, high-speed VIS and IR imaging provides a structural and thermal information about the proceeding freezing front with milisecond resolution. On the computational side, molecular dynamics simulations provide an atomistic picture of the initial state of ice nucleation at the sub-microsecond timescale. Homogeneous ice nucleation in salty water has been successfully simulated for the first time! The below journal cover graphically depicts the new concept of "filming" ice nucleation and freezing.

Bauerecker S., Ulbig P., Buch V., Vrbka L., Jungwirth P.:
Monitoring ice nucleation in pure and salty water via high speed imaging and computer simulations.
Journal of Physical Chemistry C 112: 7631-7636 (2008).
Is the surface of neat water ion-free, neutral, basic, or acidic? In our recent PNAS
study (see also articles in Chemistry World and C&E News) we show that the
surface monolayer is actually "acidic" with "pH" about 4.8 and "pOH" around 8. (We operationally define surface "pH" or "pOH" as the negative logarithm of hydronium or hydroxide
concentration in the top-most layer being aware of the fact that true pH, defined via hydronium activity, is the same at the surface as in the bulk.) We base this conclusion on
ab initio and classical MD simulations of the ionic product of water, spectroscopic experiments, as well as on previous computational and experimental studies
showing surface propensity of hydronium ions. This result can be relevant for aqueous systems with large surface to bulk ratio, such as microscopic atmospheric
aerosols. The picture below shows a snapshot from a MD simulation with surface bound hydronium and bulk hydroxide.

Buch V., Milet A., Vácha R., Jungwirth P., Devlin J.P.:
Water surface is acidic.
Proceedings of the National Academy of Sciences 104: 7342–7347, 2007.
October 2006
Our study on the higher affinity of sodium over potassium to protein surface has appeared in PNAS. The results, which are pictorially shown below (sodium: green balls, potassium: blue balls, protein: RNase A), may provide hints as to why we are burning about 30 % of our available energy (1 meal per day!) to pump sodium out of the cell.
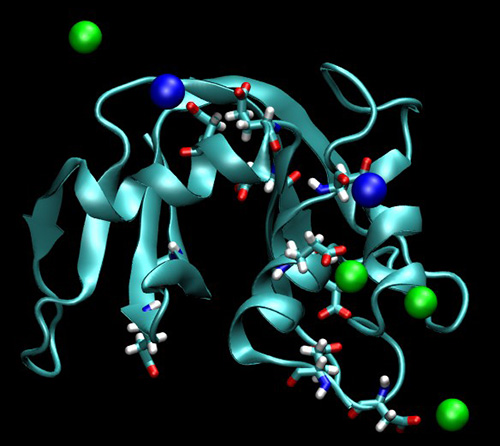
Vrbka L., Vondrášek J., Jagoda-Cwiklik B., Vácha R., Jungwirth P.:
Quantification and rationalization of the higher affinity of sodium over potassium to protein surface.
Proceedings of the National Academy of Sciences 103: 15440-15444, 2006.
October 2006
We have also looked into the onset of dissolution of complex salts recently. The below cover of PCCP shows the step-by-step hydration of NaSO4⊖ by water molecules, as investigated by photoelectron spectroscopy and quantum chemical methods. Our ab initio calculations provide a detailed picture of the build up of the hydration shell and transition from a contact ion pair to a solvent separated ion pair.

Wang X.-B., Woo H.-K, Jagoda-Cwiklik B., Jungwirth P., Wang L.-S.:
First steps towards dissolution of NaSO4⊖ by water.
Physical Chemistry Chemical Physics 37: 4294-4296, 2006.
September 2006
We have succeeded to freeze a slab of water from scratch. It is trivial to do it in the fridge but try it on the computer! Below is a JPCB cover showing that homogeneous ice nucleation starts preferentially in the subsurface. This has important implication for the microphysics of cirrus and polar stratospheric clouds and, consequently, for the global radiative balance of the Earth.

Vrbka L., Jungwirth P.:
Homogeneous freezing of water starts in the subsurface.
Journal of Physical Chemistry B 110: 18126-18129, 2006.
June 2006
Karl Popper showed us that nothing can be proved in science (only falsified). OK, nevertheless, here we show results from our simulations indicating the presence of iodide (but not fluoride or sodium) at the surface of water (but not methanol), supported by Metastable Impact Electron Spectroscopy. There is of course a small catch - simulations were done in water, while the experiment in glassy amorphous solid water, however, both have very similar structural properties.
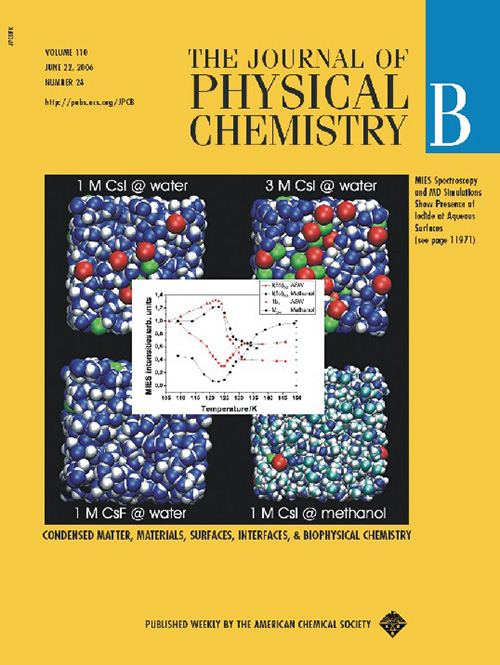
Hoefft O., Borodin A., Kahnert U., Kempter V., Dang L.X., Jugwirth P.:
Surface segregation of dissolved salt ions.
Journal of Physical Chemistry B 110: 11971-11976, 2006.
April 2006
A good part of our efforts is directed towards elucidating the behaviour of ions at the air/water interface by a pragmatic combination of molecular dynamics simulations and ab initio quantum chemistry calculations. This study, which puts in question the traditional model of an ion-free surface of aqueous electrolytes, has also direct atmospheric implications (e.g., for the chemistry of aqueous sea salt aerosols or for thundercloud electrification). With Barbara Finlayson-Pitts and Doug Tobias we have put together a special issue of Chemical Reviews dedicated to structure and chemistry at aqueous interfaces, which summarizes recent computational and experimental findings. The below cover shows the active role of ions at the solution/wapor interface.

Jugwirth P., Tobias D.J.:
Specific Ion Effects at the Air/Water Interface.
Chemical Reviews 106: 1259-1281, 2006.
May 2005
Multiply charged ions behave differently since the "bulk driving" electrostatic force is much stronger than for monovalent ions and overwhelms the "surface driving" polarization interactions. A good example is the sulfate dianion, which is very strongly repelled from the air/water interface. This is demonstrated on a 2005 Journal of Physical Chemistry B cover showing the ion-free surface layers of aqueous sulfate salts, which is also manifested in the experimental VSFG spectra.
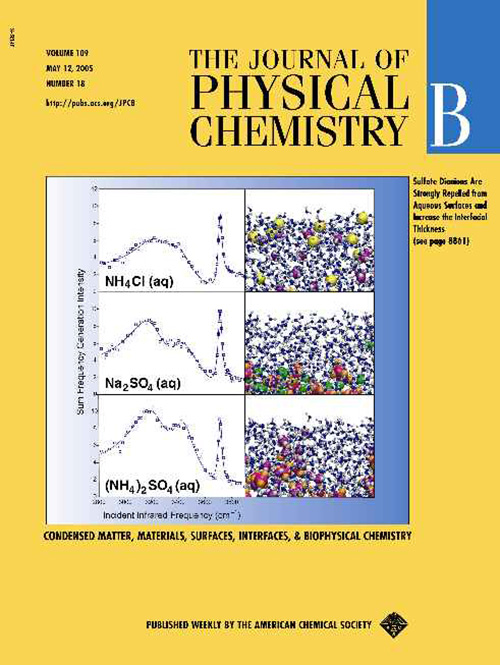
Gopalakrishnan S., Jungwirth P., Tobias D.J., Aleen H.C.:
Air-Liquid Interfaces of Aqueous Solutions Containing Ammonium and Sulfate: Spectroscopic and Molecular Dynamics Studies.
Journal of Physical Chemistry B 109: 8861-8872, 2005.
April 2005
This JPCB cover summarizes our reasearch demonstrating the different structure of the surface of aqueous acids, bases, and salts: In strong monovalent inorganic acids (such as HCl, HBr, or HI) both cations (hydronium) and anions exhibit propensity for the interface. There is a net accumulation of ions n the interfacial layer and, consequently, these acids reduce surface tension of water. In inorganic salts (such as NaCl and other alkali halides) and bases (such as NaOH) the cations are repelled from the surface while the anions exhibit a varying surface propensity (often enhanced at the surface and depleted in the sub-surface), depending on their polarizability, size, and other properties. As a result, there is a net depletion of ions from the interfacial layer and, consequently, these salts and bases increase the surface tension.
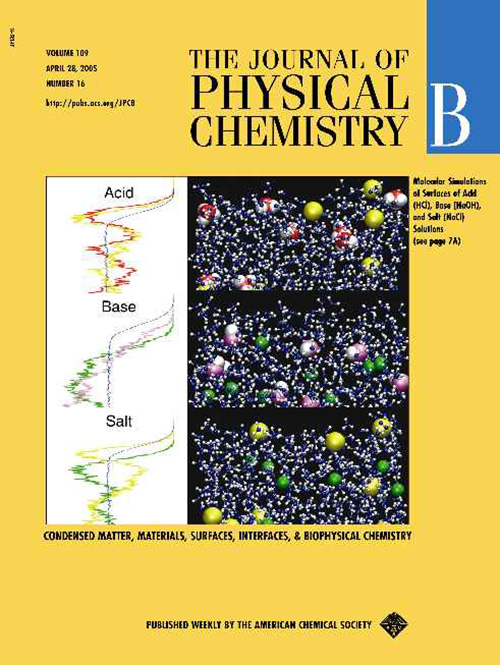
Mucha M., Frigato T., Levering L., Allen H.C., Tobias D.J., Dang L.X., Jungwirth P.:
Unified Molecular Picture of the Surfaces of Aqueous Acid, Base, and Salt Solutions.
Journal of Physical Chemistry B 109: 7617-7623, 2005.
August 2004
Below is the cover page of a 2004 Australian Journal of Chemistry issue showing our simulations of ionic surfactants: aqueous tetra-butyl ammonium fluoride with cations at the solution/vapor interface.

Vrbka L., Jungwirth P.:
Counter-Ion Effects and Interfacial Properties of Aqueous Tetrabutyl Ammonium Halide Solutions.
Australian Journal of Chemistry 57: 1211-1217, 2004.
June 2002
Below is the cover page of a 2002 Journal of Physical Chemistry B issue containing our Feature Article "Ions at the Air/Water Interface" which summarizes our results on the propensity of heavier halides (chloride, bromide, and iodide) for the air water interface.

Jungwirth P., Tobias D.J.:
Ions at the Air/Water Interface.
Journal of Physical Chemistry B 106: 6361–6373, 2002.